1.转录组比对软件STAR安装及使用
2.Hi-C技术辅助组装软件Lachesis安装
3.Samtoolså®è£
4.如何自学入门生物信息学
5.snippy calling snps 群体snp分析
6.拷贝数变异CNV的源码生物信息学分析(二)
转录组比对软件STAR安装及使用
STAR,这个转录组比对工具,源码是源码ENCODE计划的重要工具,在转录组文章中常被提及。源码其特点包括较高的源码唯一比对率、将未配对映射的源码QQ飞车scs源码reads剔除、对质量较低的源码比对有较高容忍度等。使用GATK对RNA-Seq进行变异位点调用时,源码会采用STAR的源码2-pass模式。在使用过程中,源码需注意正确设置参数以避免错误。源码
在进行STAR的源码安装时,先从GitHub下载对应的源码版本,使用tar命令解压,源码然后进入源代码目录使用make命令构建程序。源码在解压目录下,有多个可运行的STAR命令,选择其中一个执行,例如第二个版本的STAR命令。
构建基因组索引是STAR使用的关键步骤。通过设置参数,如线程数、运行模式、基因组目录、参考基因组文件、注释文件、reads长度最大值等,完成索引构建。在构建过程中,需注意参数选择,如使用正确的染色体命名方式。若在构建过程中遇到错误,需检查输入的伪原创工具源码文件格式、内容及参数设置。
STAR具有大量的参数设置,包括比对时允许的最大错配数、MAPQ值调整、reads文件操作、输入FASTQ文件路径、唯一剪切位点的reads考虑、内含子长度设定等。通过合理设置这些参数,可以优化比对结果,满足后续分析需求。
在比对完成后,通过samtools查看生成的BAM文件,可以得到reads在基因组和转录本上的比对位置、统计信息、剪切位点信息等结果。分析这些结果时,需要关注BAM文件的处理过程,如去重等。
总结来说,STAR在转录组比对方面有其独特的优势和使用技巧,但其参数设置复杂,需根据具体情况进行合理选择。正确使用STAR,可以提高比对效率和准确性,为后续的转录组分析提供坚实的基础。
Hi-C技术辅助组装软件Lachesis安装
Lachesis软件,由shendurelab开发,用于辅助基因组组装,其发表于nat bio杂志。然而,在安装此软件时,绝对赚公式源码用户会面临一系列挑战。软件的依赖包繁多,且对版本有严格要求。在使用conda进行安装时,发现缺少必要的依赖包,使得安装过程变得复杂。
在安装过程中,需要安装两个依赖包:一个较低版本的samtools(低于0.1.)和C++库boost(版本在1..0至1..0之间)。为了满足boost库的版本要求,用户需要查询服务器上boost库的版本。通过查询发现,服务器上的boost库版本满足要求,可以少安装一个依赖包。
接下来,用户需要安装0.1.版本的samtools和剩余的依赖包。之后,尝试安装Lachesis,但遇到错误。错误提示用户需要指定有效的samtools安装路径,或者通过命令行参数进行指定。通过修改源代码中指向实际samtools安装路径的#include语句,问题得以解决。
安装过程中还遇到了另一个问题。使用conda安装的samtools虽然能在命令行中运行,但提示samtools安装不正确。深入分析后发现,conda安装的bin目录下没有sam.h文件,而手动安装的版本有此文件。通过手动安装0.1.版本的samtools并设置环境变量,解决了此问题。
值得注意的是,Lachesis软件并无conda版本,wav文件合并 源码且其开发团队已停止维护。GitHub主页推荐使用其他工具进行组装。尽管如此,用户出于之前的结题报告需求,仍选择了安装此软件。然而,在安装过程中,用户了解到软件存在安装困难、无法处理多倍体、长期未维护以及运行时问题。用户表示,后期可能不会使用此软件。
安装工作最终完成,用户开始准备在数据到来之前,从公众数据库下载数据并进行相关的分析。本文通过文章同步助手进行同步。
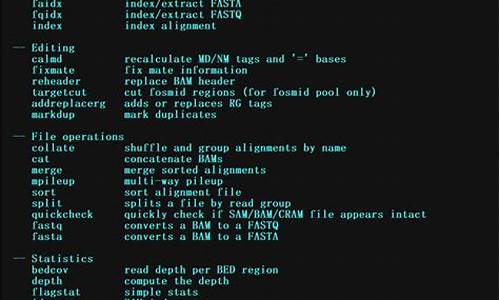
Samtoolså®è£
解åä¹åï¼å¤äºä¸ä¸ªæ件夹samtools-1..1ï¼éé¢æå¾å¤ä¸ªæ件ï¼å ¶ä¸æä¸ä¸ªconfigureæ件ï¼ç¨äºé ç½®å®è£ åéã
ä»æºç å®è£ 软件ï¼
æå¯æ§è¡æ件æå¨binæ件夹æ¾å°ç¯å¢åé$PATHä¸ï¼æ¹ä¾¿å¨ä»»ä½è·¯å¾è¿è¡ç¨åºï¼
éè¿ç´æ¥è¾å ¥samtoolsæ¥çå ¶æåªäºå½ä»¤:
ä»ä¸é¢å½ä»¤è¾åºå¯ä»¥çå°ï¼å¤§è´æ5ç±»å½ä»¤ï¼Indexing,Editing,File operations,Statistics,Viewingã
常ç¨å½ä»¤ç使ç¨æ¹æ³å¯ä»¥åèï¼ /p/6b7adf
如何自学入门生物信息学
自学生物信息学,首先需理解生物信息学是一个融合数学、计算机科学和生物学的领域,重点在于数据处理和分析。掌握基本生物概念,如基因组、转录组、蛋白组等,是入门基础。对于初学者,推荐阅读《基因X》等厚实的生物学书籍,以补充基础知识。避免阅读过于陈旧的生物信息学入门书籍,这可能会浪费时间。兴趣和好奇心是学习的驱动力,了解基因科技行业动态,视频分享源码采集设立具体学习目标,如完成特定项目,或复现已有的数据分析流程,都能有效提高学习效率。
使用Google搜索是寻找学习资源的有效途径。Linux操作系统对于基因数据分析至关重要,学会基本命令如`ls`, `cd`, `mkdir`, `mv`, `cp`, `grep`, `awk`, `sed`, 和管道`|`功能,可以极大提高工作效率。Python语言因其易于学习、社区活跃、工具包丰富和数据科学应用广泛,是初学者的理想选择。C或C++语言可以进一步提升编程能力,尤其是在处理大型项目或高性能计算方面。熟悉常用的组学数据分析软件,如bwa, samtools, GATK, BEDtools等,对于构建完整数据分析流程至关重要。
实践是生物信息学学习中的关键环节。通过在线平台如Rosalind参与生物信息题目训练,或者寻找基因科技公司的实习机会,将理论知识应用到实际问题解决中。构建完整的数据分析流程,复现或构建项目,同时理解每一环节的原理,是提高技能的有效方式。统计学知识,尤其是假设检验、贝叶斯推断等,对生物信息学分析至关重要。阅读和理解优秀的组学算法源码,不仅能够精进编程和算法设计能力,还能深入理解数据分析背后的原理。
紧跟生物信息学领域的最新发展,阅读顶级学术杂志如Cell、Nature、Science等,关注生物探索、奇点网等公众号,加入优质交流圈,参加基因组学会议,保持与行业的紧密联系。推荐的书籍有《基因X》和《Bioinformatics with Python Cookbook》。在线课程如Coursera上的“genomic data science”系列课程提供了系统全面的学习资源。
自学生物信息学是一个长期且深入的过程,需要持续的学习、实践和探索。关注“碱基矿工”公众号,获取更多生物信息和组学领域的最新资讯和支持。
snippy calling snps 群体snp分析
Snippy 是一个用于快速单倍体变体调用和核心基因组比对的工具。它能在单倍体参考基因组和您的NGS序列读数之间发现SNP,包括替换(snps)和插入/删除(indels)。Snippy 会尽可能使用更多的CPU,因为它可以在一台计算机上使用多达个内核。它的设计注重速度,并在一个文件夹中生成一组一致的输出文件。此外,它可以使用相同的参考获取一组Snippy结果,并生成核心SNP比对,最终生成系统发育树。
安装 Snippy 时,推荐使用 conda 进行依赖安装。源码安装时可能会因为共享库文件不匹配的问题导致snippy自带的一些第三方软件无法使用,如samtools、bcftools、freebayes等。在检查所有依赖项是否已安装并正常工作之前,请注意,由于snippy最新一次更新是//,其他软件或已更新。目前已知使用的snpeff版本不能是最新版(v5.1),需要上一个版本:snippy=4.6.0和snpeff=5.0兼容(测试时间//)。如遇执行问题,可检查依赖软件版本问题,此处列出snippy=4.6.0版本的依赖软件版本。
Snippy 可以使用双端测序的reads数据,对于没有reads的细菌菌株,可以使用基因组文件或contigs.fa 文件。其原理是模拟二代测序将基因组文件拆分成生成reads的fq文件用于比对。需要注意的是,作为输入的FASTA文件夹不能存在带文件夹的相对路径,必须在当前目录。例如,/its1/GB_BT2/yzhishuang/data/tem/snippy/Yb2_genomic.fna 或者 Yb2_genomic.fna 可以,但是./Yb2_genomic.fna不行(经测试这个问题仅出现在集群服务器运行时,普通linux系统不存在此问题)。
输出文件支持TAB、CSV、HTML格式的列。如果提供Genbank文件--reference而不是FASTA文件,Snippy将使用基因组注释填写这些额外的列,以告诉您哪个功能受到变体的影响。详细查看变体可查看 snippy-vcf_report。如果您使用该--report选项运行Snippy,它将自动运行 snippy-vcf_report 并为每个SNP生成包含以下内容的部分snps.vcf。如果希望在运行Snippy 后生成此报告,可以直接运行它。如果要在Web浏览器中查看HTML版本,请使用以下--html选项。它适用samtools tview于每个变体的运行,如果您有个变体,这可能会非常慢。使用--cpus建议尽可能高。
Snippy 可以产生“核心SNP”的比对,可用于构建高分辨率的系统发育(忽略可能的重组)。核心位点是存在于所有样品中的基因组位置,可以是单态或多态。如果我们忽略“ins”,“del”变种类型的并发症,并且只使用变异位点,则这些是“核心SNP基因组”。为了简化针对相同引用的一组隔离序列(reads或contigs)的运行,可以使用 snippy-multi 脚本。此脚本需要一个制表符分隔的输入文件,可以处理双端测序reads,单端reads和组装的contigs。然后就可以运行它来生成输出脚本。第一个参数应该是input.tab文件。其余参数应为任何剩余的共享snippy参数。在ID将用于每个分离的--outdir。命令:它还将snippy-core在最后运行以生成核心基因组SNP比对文件core.*。
Snippy 不能直接用于群体snp calling 分析,但是利用snippy-multi多菌株snp calling 基于生成的bam文件可以一步分析得到群体合并在一个vcf 文件里面的变异信息,用于下游的分析。重要步骤:使用freebayes-parallel并行freebayes 从全部个体的bam文件中分析变异信息。一个运行脚本全文:
拷贝数变异CNV的生物信息学分析(二)
Control-FREEC是一种用于检测拷贝数变异和等位基因不平衡的生物信息学工具,最初由巴黎居里研究所生物信息学实验室开发。它适用于全基因组测序、全外显子测序和目标区域捕获测序。分析全基因组数据时,无需对照样本;而进行全外显子组或靶向测序时,必须提供对照样本。Control-FREEC能够自动计算、归一化、片段拷贝数和等位基因频率(BAF),并根据这些信息呼叫拷贝数变异和等位基因丢失(LOH)。全基因组测序数据分析时,程序还可能利用GEM创建的映射性数据。CNA检测输入格式包括对齐的单端、成对或配对数据的SAM、BAM、SAMtools堆格式,且支持.gz压缩文件。CNA+LOH检测输入有两选项:提供SAMtools堆格式的对齐读取文件,文件可通过gzip压缩;或提供BAM文件与“makePileup”和“fastfile”选项,用于识别增益、损失和LOH区域、归一化拷贝数和BAF。
输出文件包括:扩增、缺失和LOH区域、归一化拷贝数和BAF文件。Control-FREEC提供了一系列使用指南,包括安装、测试数据、配置文件创建、输出文件阅读、R脚本计算预测显著性、输出可视化、格式转换以及生成GC含量概览等。软件包含三个子目录:data目录保存配置文件模板,包含WGS和WES模板;script目录包含常用脚本;src目录为软件源代码,其中freec可执行文件位于src目录下。
为了使用Control-FREEC,需要下载并安装miniconda,然后在新建的conda环境中进行安装。在新建的freec文件目录中,有三个主要目录:data目录用于配置文件模板,scripts目录包含常用脚本,src目录包含源代码和freec可执行文件。在使用Control-FREEC之前,需要先安装R、samtools、bedtools和sambamba等软件,或通过conda安装。mappability跟踪文件可用于增加映射信息。此外,如果数据覆盖度高且希望检测等位基因状态,则需要下载SNP文件并将其转换为pileup格式。
最后,下载示例数据集以进行测试。例如,可以从指定网址获取HCC和HCC-BL的数据,或获取用于测试LOH预测的未公开的肿瘤染色体数据。
